 |
 |
 |
 |
 |
In the 21st century, one of the most important problems facing humanity is developing an enduring, sustainable energy economy. The Bartlett group uses inorganic synthesis to prepare compositionally complex materials—materials in which multiple specific functions such as light harvesting, charge separation, and charge transfer are required, but each component can be tuned to optimize a single function. A central theme of our program is the interplay between structure, composition, and physical properties such as optical absorptivity and electrical conductivity. We aim to control morphology and composition in nanoscale materials with two main thrusts: solar energy conversion to chemical fuels in light-harvesting metal oxides and electrical energy storage in ion-insertion battery materials.
Solar Energy Conversion
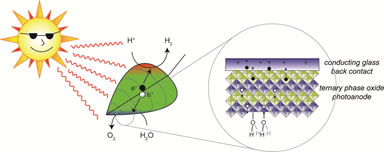
Crystalline inorganic semiconductors are extensively studied as photocatalysts for fuel-forming reactions such as water splitting. The key limitation toward employing semiconductor photocatalysts for solar fuels production is finding a material that meets four stringent requirements: 1) it strongly absorbs visible light; 2) the collection efficiency of photogenerated charge carriers is high; 3) it is chemoselective for water oxidation in the presence of dissolved ions such as chloride in sea water); 4) it is chemically robust under prolonged solar illumination. The first example of photoelectrochemical water splitting was carried out on TiO2, but this material only absorbs in the ultraviolet part of the solar spectrum (UVA rays), and the solar flux drops off at shorter wavelengths (TiO2 is a classic sunscreen). The common non-oxide III-V and II-VI semiconductors such as GaAs and CdS that are common visible-light solar cell materials corrode in water; under illumination, the As3– or S2– anions are oxidized over water preferentially. Two other inexpensive workhorse materials for water oxidation are Fe2O3 and WO3, whose electronic structures are such that the electrons excited from valence- to conduction band are not sufficiently reducing to generate H2 from water. A growing focus on separating oxygen evolution from hydrogen evolution alleviates this drawback; this is akin to how green plants separate oxygen evolution from reducing carbon dioxide in natural photosynthesis. However, photogenerated charge carriers move too slowly in Fe2O3, and WO3 is only stable under strongly acidic conditions (it dissolves from the electrode surface at neutral or basic pH). The downside of working in strong acids is that the overpotential needed to oxidize water at any appreciable rate is larger. Our group is primarily concerned with the more thermodynamically difficult and more kinetically demanding 4-proton-4-electron water oxidation half reaction to liberate oxygen. The central hypothesis of our work is that compositionally complex (3 or more element-containing) materials orthogonalize the thermodynamics of water oxidation (optical band gap and oxidizing potential) and the reaction kinetics (rate of oxygen evolution).
Over the past several years, we have synthesized and characterized ternary materials in two structural classes, anatase and wolframite, that demonstrate excellent chemical stability during photocatalysis. We began this work with two goals in mind:
-
Goal 1: To red shift the absorption of TiO2 into the visible part of the solar spectrum by co-alloying with charge-compensated ion pairs.
Here, our key contributions include: 1) synthesizing the first example of a co-incorporated anatase compound, TiO2:(Nb,N) with visible light electronic transitions; 2) demonstrating visible light generation of active oxygen species needed for photocatalysis, with water oxidation as a specific example.
-
Goal 2: To generate tungsten oxide-based photoelectrodes that are stable in neutral and basic conditions by preparing insoluble transition-metal tungstate salts.
Here, our key contributions include: 1) preparing CuWO4 photoanodes by electrochemical deposition and sol-gel processing; 2) demonstrating for the first time oxygen evolution on CuWO4 with concomitant ferricyanide reduction using only simulated solar radiation (no applied electrical bias); 3) demonstrating chemoselectivity for water oxidation on CuWO4 in saline buffered solutions; 4) showing that we can prepare mixed-metal solutions by straightforward sol-gel synthesis routes.
Electrical Energy Storage
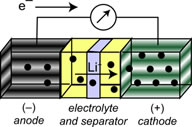
Li-ion insertion materials are common to our everyday experience with batteries in portable electronics, power tools, and electric vehicles. Li-ion technology is a mainstay because lithium is the lightest metal on the periodic table and offers the highest voltages. In lithium-ion batteries, the primary research challenges are suppressing deleterious side reactions at solid-liquid interfaces for long-term stability, and designing compositions and/or morphologies of materials that allow for rapid insertion and extraction of lithium ions to charge and discharge batteries at reasonable rates. Our work focuses highly on the spinel class of materials, and the spinel LiMn2O4 (LMO) has keen commercial relevance: it comprises the cathode in the battery of the Chevy VoltTM plug-in hybrid electric vehicle. Manganese in the battery is cycled between formal oxidation states +3 and +4, which is simultaneously advantageous and detrimental. The advantage is that the excess electron inserts in and is extracted from a strongly Mn–O σ-antibonding orbital, which gives rise to its large voltage (+4.1 V vs. Li+/0). However, the Mn3+ ion is subject to a structural (Jahn-Teller) distortion that, propagated over the entire cubic crystal, results in mechanical strain and ultimately cell failure by crystallite fracture. A second problem is that Mn3+ spontaneously disproportionates (2Mn3+ → Mn4+ + Mn2+), and Mn2+ on the surface is soluble in the electrolyte solution. Manganese metal can then plate on the anode and provide reaction sites for the solvent to be reduced, building a larger solid-electrolyte interface (SEI) layer that can retard Li+ insertion into the anode (typically graphite). Then, graphite itself can undergo side reactions with Li+ to plate high surface area Li-metal dendrites that can react with the solvent, electrolyte, or separator materials. In the mid-1990s this mechanism was uncovered as the culprit for laptop batteries catching fire. In our work, the central hypothesis is that nanoscale oxide materials have a larger surface-to-volume ratio that will better accommodate mechanical strain. Well-defined compositions will hinder disproportionation, and together, these features will yield safer (chemically stable) batteries with long cycle lives needed for commercial applications.
Over the past several years, we have prepared spinel-based oxides for both cathodes and anodes of batteries that demonstrate long cycle lives and rapid Li-ion diffusion. We started with two goals in mind:
-
Goal 1: To develop low-temperature methods for controlling oxygen content in spinel-phase materials.
Here, our key contributions include: 1) developing a one-pot redox hydrothermal synthesis of LMO using oxygen gas as an oxidant and simple organic molecules as reducing agents; 2 quantifying defect composition by neutron diffraction, electrochemical, and thermogravimetric methods; 3) expanding this synthetic method to generate the higher voltage nickel-containing spinel compound based on LiNi0.5Mn1.5O4 (LNMO).
-
Goal 2: To apply sol-gel methods for materials coatings to improve electrical conductivity or chemical stability of battery materials.
Here, our key contributions include: 1) synthesizing carbon-coated nanoparticles of the anode material Li4Ti5O12 that show reasonable cycling rates; 2) preparing titania coatings to prevent breakdown of the LNMO materials described above; 3) expanding hydrothermal methods for bronze-phase TiO2-B materials.