In this section we will step you through the HCN isomerization example and along the way we will explain all the available options and what they all mean superficially and in-depth.
Reactant calculations:
Step 1:
Open Spartan!
Step 2:
Click on the small white icon in the bar directly above the teal main window, it looks like this
. This will open a new sheet in the build option. The gray column on the right is where you will find all of your usual atoms along with an expert model kit that defies normal bonding rules. Also contained in the atomic building kit is a section with proteins and nucleic acids. Let's go ahead and stay in the normal section for now.
Now click on the button that is labeled with a triple bond carbon and then click in the teal work area. Next add a triple bond nitrogen by first clicking on the button then on the triple bond of the carbon atom. Then add a hydrogen atom to the remaining free orbital.
Now we have our molecule!
It should look something like this:
Without the labels.

Step 3:
Click on setup in the drop down menu bar and select calculations...
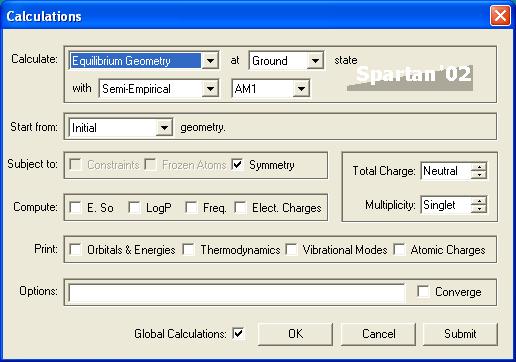
This is what you should get. Under the section Calculate there are several options available:
Let's go ahead and explain all of these options:
Single point energy : This calculates energies and thermodynamic quantities using the structure that is drawn in the work space. For this calculation the molecule's geometry is not changed or optimized before the final calculation.
Equilibrium Geometry : This calculation first optimizes the geometry of the molecule and then calculates the energy and thermodynamic quantities. Geometry optimization finds the molecular confirmation with the lowest energy. This process involves moving each individual atom in the molecule in small steps in all directions to test whether the energy increases or decreases. In the event of an energy increase the program knows that the initial state was more energetically favorable, and if the energy decreases the new molecular confirmation is more energetically favorable. In the end the molecular confirmation with the lowest energy is used in the final calculations.
Transition State Geometry : This calculation finds the transition state of the reaction. The transitions state is found by first finding a high energy conformation of the molecule or molecules that also has a imaginary frequency which is characteristic of a transition state. After the transition state geometry is found the energy and thermodynamic quantities are calculated.
Equilibrium Conformer : This function finds additional equilibrium geometries by moving each atom in fairly large spatial steps, and displays the most energetically favorable conformation.
Conformer Distribution : This function like the previous function finds all possible molecular conformations but unlike the previous function displays a list of all conformers.
Energy Profile : This function will calculate energies at a user specified number of points along a user defined molecular movement. In other words it will generate a intrinsic reaction coordinate for a reaction. This function is slightly tricky to use, but when it works it is a nice addition to your data set.
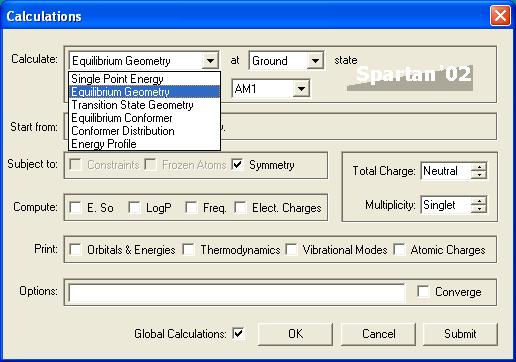
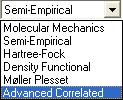
For our first run choose the equilibrium energy calculation and notice the drop down box directly below the function selection drop down box. This drop down menu contains different calculation techniques that all produce roughly the same results with differing accuracy.
Click here for an in-depth explanation of these calculation methods
As you move down the list computation time increases rapidly. For now we will leave the calculation method as semi-empirical.
The drop down menu directly next to the calculation selection drop down menu is an option to do the calculations either at ground state or at an excited state (i.e., the calculation will be done at the first excited state).
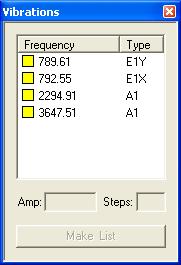
In the compute section check the Freq. box and under the print section check the box for thermodynamics, these selections will compute the frequencies of vibration for the molecule that can in turn be used to calculate thermodynamic quantities.
Step 4:
Click submit.
Step 5:
Check the output by clicking display in the menu bar and select output. The output should look something like this:
Spartan '02 Mechanics Program: (PC/x86) Release 116
Heat of Formation: 31.007 kcal/molZero-point vibrational energy is 10.951 kcal/mol
Standard Thermodynamic quantities at 298.15 K and 1.00 atm
Translational Enthalpy: 0.889 kcal/mol
Rotational Enthalpy: 0.592 kcal/mol
Vibrational Enthalpy: 11.008 kcal/mol
....Total Enthalpy: 12.489 kcal/molTranslational Entropy: 35.816 cal/mol.K
Rotational Entropy: 11.823 cal/mol.K
Vibrational Entropy: 0.231 cal/mol.K
....Total Entropy: 47.870 cal/mol.K
Free Energy (H-TS): -1.784 kcal/mol
As you can see the calculations yield the heat of formation, zero point energy (ZPE), enthalpy, entropy, and Gibbs free energy all at STP. Also of interest is the heat of formation while using the semi-empirical calculation setting this is the overall energy of the molecule. Now if we were using one of the other more complicated calculation settings the heat of formation would be replaced with the ground state energy (GSE).
Let's run the same calculation using density functional instead of semi-empirical and see what kind of numbers we get. Just simply open the calculation window again and change the calculation settings and leave everything else the same. Then click submit.
Spartan '02 Mechanics Program: (PC/x86) Release 116
Optimization:
Step Energy Max Grad. Max Dist.
1 -93.4213658 0.048177 0.058300
2 -93.4226115 0.004842 0.004136
3 -93.4226273 0.000311 0.000408
4 -93.4226273 0.000004 0.000007Molecule is linear
Zero-point vibrational energy is 10.338 kcal/molStandard Thermodynamic quantities at 298.15 K and 1.00 atm
Translational Enthalpy: 0.889 kcal/mol
Rotational Enthalpy: 0.592 kcal/mol
Vibrational Enthalpy: 10.449 kcal/mol
....Total Enthalpy: 11.930 kcal/molTranslational Entropy: 35.816 cal/mol.K
Rotational Entropy: 11.815 cal/mol.K
Vibrational Entropy: 0.472 cal/mol.K
....Total Entropy: 48.103 cal/mol.KFree Energy (H-TS): -2.412 kcal/mol
Comparing enthalpy, entropy, heat of formation, and ground state energy:
|
Method |
Enthalpy |
Entropy |
Heat of Formation |
Ground State Energy |
|
Semi-emperical |
12.489 kcal/mol |
47.870 cal/mol |
31.007 kcal/mol |
----- |
|
Density Functional |
11.930 kcal/mol |
48.103 cal/mol |
----- |
-93.423 au -58622.933 kcal/mol |