In this section we will step you through the HCN isomerization example and along the way we will explain all the available options and what they all mean superficially and in-depth.
Reactant calculations:
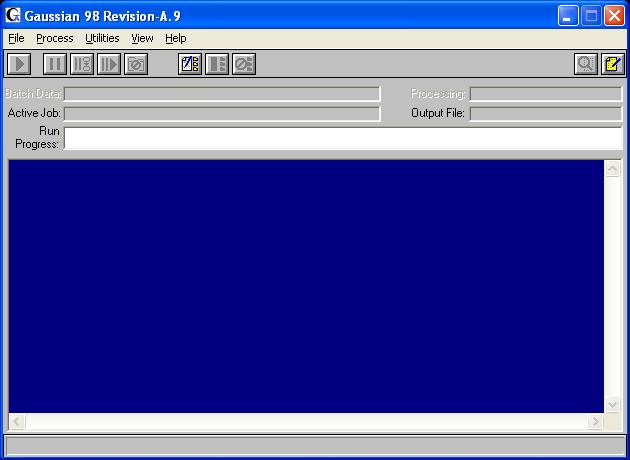
Open Gaussian. You should see a window that looks something like this:
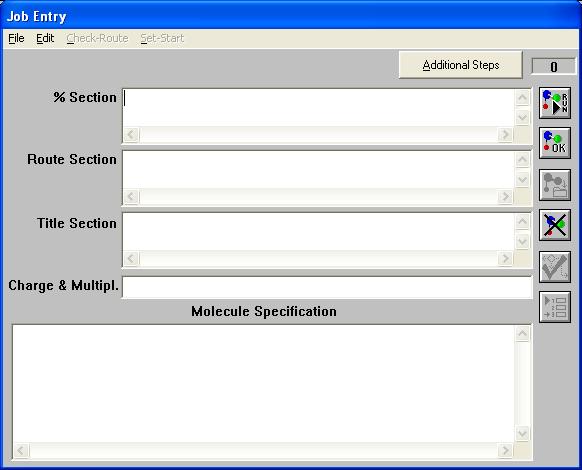
Click on File and select New. Now a window that looks like this should pop up:
This is the input window for Gaussian, the first input is the Link zero commands. The Link zero commands are commands that tell Gaussian what kind of additional files to create and what information to input and retrieve from these additional files. For our purposes we will not be needing this section of the input window. Next, the Route Section is where the type, method, and specification of the calculation will be specified. The Title Section is where you will input the title of your calculation, this section is a place holder and must be included in the Gaussian input. Charge and Multiplicity is specified next. Charge is the overall charge of the molecular system and multiplicity describes how the electrons of the system exists. Multiplicity is mathematically defined as 2S+1 = spin multiplicity, where S equals the total angular momentum of the unbound electrons. Finally is the Molecule Specification, where the geometry of the molecular system is defined.
Lets go ahead and set up the calculation. First, in the Route Section type "#T RHF/6-31G Opt Freq" without the quotes. The pound sign designates this line as a command line. The " T " tells Gaussian to suppress unnecessary output. The " U " specifies that the electron shell of the molecule is unrestricted. " HF " specifies that the Hartree Fock calculation method is to be used. " 6-31G " specifies the basis set to be used. 6-13G is a popular basis set that approximates the electronic structure of an atom with six Gaussian distribution functions for the core orbital, the valence orbital is then represented by two functions, one that is a set of three Gaussians and a second function that is a single Gaussian function. " Opt " tells Gaussian to perform a geometry optimization on the molecules specified in the system. " Freq " tells Gaussian to perform a frequency calculation on the HCN molecule.
Next give your calculation a name (i.e. hcn reactant geo opt freq), and input a charge and a spin multiplicity separated by a space or a comma. The spin multiplicity = 2S+1 where S is the total angular momentum of the molecule. Try to figure out the charge and the spin multiplicity and click here to check your input.
Now we need to input the geometry of the molecule. The easiest way to specify the HCN molecule in Gaussian is to use internal coordinates. When using internal coordinates there are four types of input, type of atom, atomic distance, angle produced by three atoms, and dihedral angles formed by four atoms. The general input format is as such:
- X
- X,1,D
- X,2,D,1,A
- X,3,D,2,A,1,DHA
Now click on the Run button,
, and save the output file to the scratch drive D:. The calculation steps are shown in the Gaussian main window displayed in real time. The calculation will take around 20-30 seconds on a 2.5 GHz processor. After the calculation is finished you can view the output by clicking the view button,
.
The output should look very similar to this:
SCF Done: E(UHF) = -92.8751974300 A.U. after 1 cycles
Temperature 298.150 Kelvin. Pressure 1.00000 Atm.
Zero-point correction= 0.017992 (Hartree/Particle)
Thermal correction to Energy= 0.020465
Thermal correction to Enthalpy= 0.021410
Thermal correction to Gibbs Free Energy= -0.001323
Sum of electronic and zero-point Energies= -92.857205
Sum of electronic and thermal Energies= -92.854732
Sum of electronic and thermal Enthalpies= -92.853788
Sum of electronic and thermal Free Energies= -92.876520
Open the output file with Windows notepad and click on the edit drop down menu and select find. The perform a search for "E(UHF)" without the quotes. Click on find next untill you have found the last entry with E(UHF). This last entry is the groundstate energy. Record this number. A little further down in the document you will find the thermodynamic qunatites that you need to record.
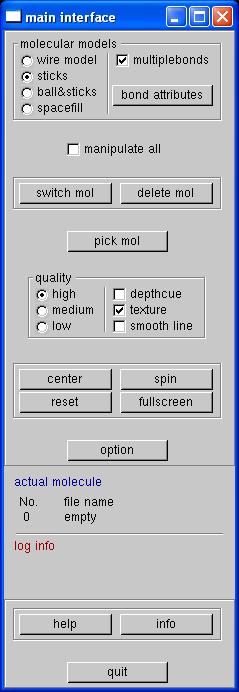
Go ahead and open Molekel, Molekel is molecular veiwing program that can use our Gaussian output to draw the HCN molecule for us.
After you open Molekel you should see the main interface window open and the Molekel main window, where you will view your molecule. To load your Gaussian out file, right click within the main window and select load and then gaussian log, a file browser window will open. Hit the backspace untill the .out is deleted, then type D:/ and hit enter. Now find your file and click on the file (this sometimes does not work) or type in the file name and hit enter. You should see the molecule drawn in the main window. In the main window you can change how the molecule is drawn by clicking on the radio buttons in the upper left of the main interface, ball & sticks should work nicely. Left click, hold, and move the mouse to rotate the molecule. Hold down shift, left click, hold, and move the mouse to translate the molecule. Hold down shift & ctrl, left click, hold, and move the mouse (and jump up and down on one foot) to zoom in and out on the molecule.
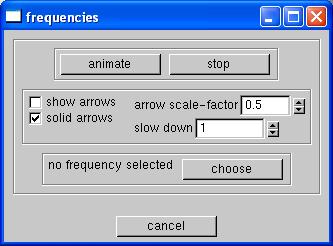
Now let's view the vibrations of the molecule. Again, right click in the main window and select animate then frequency. This will bring up yet another window that looks like this:First we need to select a frequency to animate, click on choose, and, yes, another window will open. Now select the frequency that you wish to animate and click accept. Now click on animate to view the frequency. The amplitude of the frequncy can be controled by the arrow scale-factor. To stop the animation click on ... stop. These vibrations are the natural vibrations that the molecule would be experiencing in its natural state.
Now on to the transition state calculations.
![]()
![]()