
Mass diffusivities, which represent the response of a system when a chemical potential is exerted, are essential parameters for the optimum design of various chemical or mechanical systems. However considerable uncertainties still exist especially under high temperature or high pressure conditions due to the limitation of available measurements. To overcome the limitation, Molecular Dynamics (MD) simulations combined with fully atomistic inter-molecular potential parameters are used to compute mass diffusivities.
The atomistic level potentials make it possible to consider the detailed structures of all molecules during their collisions and therefore the non-spherically symmetric interactions between polyatomic molecules can be successfully addressed. Moreover, based on the fully atomistic scale MD simulations, the modification factors, which can capture the effect of molecular structure on mass diffusivity, for the gas kinetic theory approach (Chapman-Enskog solution) can be obtained.
The goal of this project is to identify new reaction pathways for real fuels to produce a complete description of the system of interest in terms of intermediates, products, kinetics, thermochemical and transport properties. This is accomplished through the use of a combination of Molecular Dynamics and ab initio techniques.
This work is supported by theAir Force Office of Scientific Research, AFOSR.
Publications related to this project
The main objective of this work is to study the interactions of nano-systems through the development of a coarse-graining computational methodology to describe their agglomeration. This new approach allows to bridge the length and time scales in the important area of nanocluster self-assembly.
This work is supported by theNational Science Foundation.
Publications related to this project
The objectives of this project are focused on gaining a detailed biophysical knowledge of the long term interactions of nanoparticles with self-assembled biological structures, such as lipid bilayer, using Molecular Dynamics approaches. Understanding how these particles penetrate and modify the structural, thermodynamic, dynamical and mechanical properties of these biomolecular systems will help us to comprehend their potential harmful effects, as well as to develop methods for their remediation.
This work is supported by theNational Science Foundation, NSF-CAREER.
Publications related to this project
The goal of this project is to construct and improve detailed kinetic mechanisms for real biodiesel fuels by exploring new reaction pathways as well as calculating accurate data for chemical rate constant, thermodynamic, and transport properties. This is accomplished through the use of a combination of ab initio and molecular dynamics techniques. Currently, we are working on several biodiesel fuel surrogates such as methyl butanoate and methyl crotonoate. In the future, we will target on real diesel fuel molecules, e.g., methyl decanoate.
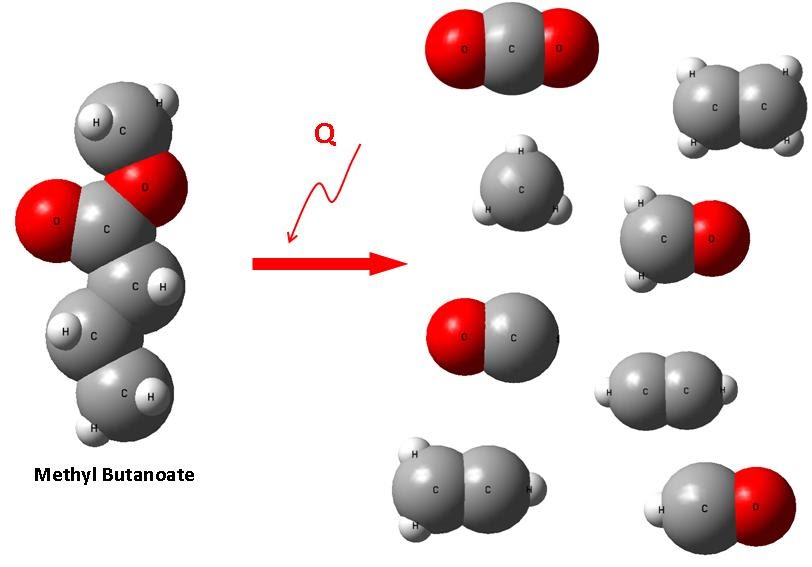
Publications related to this project
In the combustion environment, nanoparticle formation is a fascinating multiscale problem, both in length scale and in time scale. When the nanoparticles are able to reach a critical size, they begin to aggregate and the molecular science of the nanoparticles bridges with the statistical mechanics of nonequilibrium self-assembly and nucleation over two disparate length and time scales in a truly fascinating way.
The goal of this research is to develop a multiscale computational approach to characterize nanoparticle formation in combustion environments, through the use of novel simulation methodologies at disparate (spatial/temporal) regimes.
This approach provides a connection between the various time scales in the nanoparticle growth and self-assembly problem, together with an unprecedented opportunity for the understanding of the atomistic interactions underlying nanoparticle structures and growth (AMPI code- “Atomistic Model for Particle Inception” ).¹
¹ A. Violi Combustion and Flame 139: 279 (2004).
This work is supported by theNational Science Foundation.
Publications related to this project
The research foced on the development of a new reaction mechanism for novel biofules. These include biofuels like Bisabolane and Farnesane. The focus of the project was at lower temperature. Using ab initio, RRKM/ME, and VTST methods more accurately painted a picture of the reaction mechanism. Geometrical parameters, Potential energy surface, and Rate constants using Ab initio/DFT quantum chemical calculations were used to research the mechanism. Development of new Reaction mechanism for novel Biofuel molecules such as Methyl butanoate, Bisabolane and Farnesane is the final goal for the project.
© Copyright 2016 University of Michigan The Violi Group. All Rights Reserved.
Contact Us