|

|
Denitrification
The natural cycle of denitrification comprises a cascade of different enzymes that stepwise reduce nitrate
to dinitrogen [1]-[3]:

Hence, denitrification corresponds to the part of the biological
nitrogen cycle
that is opposed to nitrogen
fixation. The reduction of nitric oxide to nitrous oxide is mediated in nature by the NO reductases (NOR),
which occur in bacteria (NorBC) as well as in fungi (P450nor). The principal reaction scheme of NO reduction
corresponds to the equation:
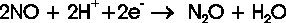
Bacterial and fungal NORs are fundamentally different in the nature of their active sites. Correspondingly,
very different reaction mechanisms have been proposed for these two classes of enzymes (see below).
In the biological denitrification cycle, nitrous oxide is reduced to dinitrogen by the N2O reductases (N2OR)
following the principal reaction scheme:
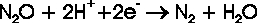
The different classes of NO and N2O reductases are described in more detail in the following sections.
Bacterial NORs (NorBC)
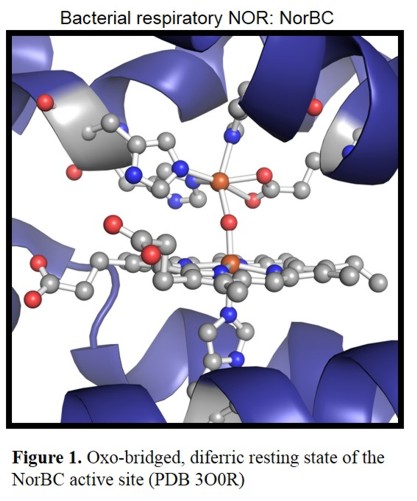
These enzymes are closely related to the respiratory heme-copper oxydases, the so called Cytochrome c-Oxidases (CCO).
Figure 1 shows the active site of the NorBC from Pseudomonas aeruginosa in the diferric resting state, which consists of a
heme b3 with axial (proximal) histidine cooedination and the so called FeB, a non-heme iron center. The latter one is coordinated by three
histidines and a glutamate.
Despite much effort, the molecular mechanism of NO reduction by NorBCs has remained largely elusive.
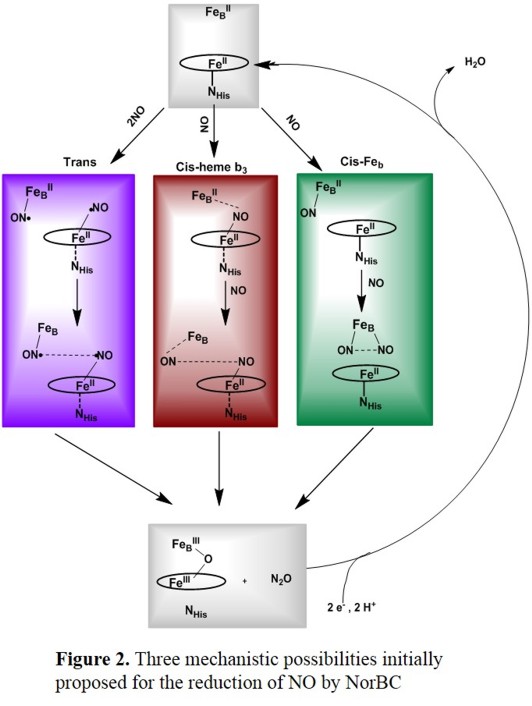
Of the many possible mechanisms that have been proposed, the ones summarized in Figure 2 represent the most likely scenarios.[5] Initial work by Girsch and de Vries favors
the trans mechanism (Figure 2, top), where catalysis starts from the diferrous form of the enzyme, and each one of the iron centers binds one NO ligand first, followed by a radical-type N-N coupling mechanism.[6]
However, heme and non-heme ferrous NO complexes as invoked in this mechanism are typically very stable and unreactive, which poses the question of how these types of complexes
could serve as reactive intermediates in the mechanism of cNORs.
In terms of the heme, reactivity of the ferrous NO complex could be increased via the trans effect in a corresponding 6C system,[7] although the radical reactivity of such complexes is still intrinsically low
as evident from corresponding diferric hyponitrite (N2O22-) complexes, which have a propensity to decompose into the corresponding ferrous heme-nitrosyls,[8]
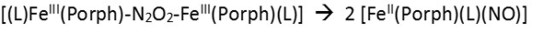
both in the absence and presence of an axial N-donor ligand L.
The cis-heme b3 mechanism in Figure 2, middle is inspired by the mechanism of Cyt. P450nor (see below), where one molecule of NO is bound first to the heme and subsequently activated for reaction with the second NO.
A variation of this pathway where the heme-bound NO interacts with the FeB center is favoured by computational methods, according to Blomberg and coworkers. In this case, the reaction would also start from
the diferrous form of the enzyme. The first NO binds to the heme, forming a six-coordinate ferrous heme-nitrosyl complex that is activated by interaction with the FeB center. The second NO then attacks the bound NO
and, at the same time, coordinates with its O-atom at the non-heme iron center. This leads to the direct generation of a bridged hyponitrite complex, whereas the non-heme iron-nitrosyl complex never forms.
According to the latest study, this mechanism is energetically favourable compared to the trans mechanism, which is disfavoured by high activation energies.[9]
The equivalent of the cis-heme b3 mechanism where the whole reaction proceeds at the non-heme iron center has also been proposed (see Figure 2, bottom). In this case, the formation of a ferrous dinitrosyl complex has
been proposed. However, no further evidence for this mechanism is available, and on the contrary, arguments can be made why this mechanism is unlikely. First, a corresponding ferrous dinitrosyl complex has never been
observed in any enzyme or model system. Second, in analogous (but more reduced) DNICs, the Fe-NO units are strongly antiferromagnetically coupled, which forces the two NO ligands to have their unpaired electrons
aligned with parallel spins. The necessary spin-flip of one NO unit required to generate an N-N bond is energetically extremely unfavourable, which constitutes a general road block for this mechanism.
This is also the reason why DNICs are generally stable, and do not show any propensity for N-N bond formation.[10]
Fungal NORs (P450nor)
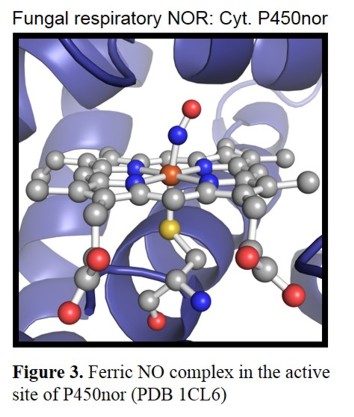
In contrast to the bacterial NORs, which are related to the CCOs, the fungal NORs are derived from Cytochrome P450 and hence,
are designated as P450nor.[11] The crystal structure of the enzyme from Fusarium oxysporum shows a heme b with the typical axial
cysteine thiolate ligand (Figure 3).[12]
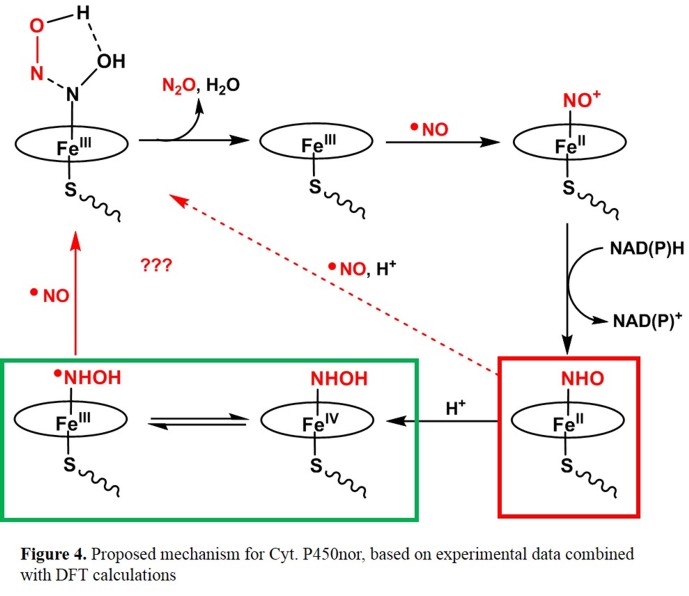
Several mechanistic studies have shown that the Fe(III) form is catalytically active in the case of
P450nor. Coordination of NO then leads to a six-coordinate low-spin complex, that has been characterized with different
spectroscopic methods. Figure 3 shows a possible mechanism, which is based on kinetic investigations [13] and DFT calculations.[14]
In this proposal, the Fe(III)-NO complex reacts with NAD(P)H in an unusual hydride transfer to generate a ferrous heme-NHO complex. Either
this species, or the corresponding, protonated hydroxylamide complex corresponds to the reactive "Intermediate I", which then reacts with a
second molecule of NO forming the product N2O. The computational results predict that the imitially formed HNO complex is basic (due to
the presence of the axial cysteinate ligand, and that Intermediate I in fact corresponds to the protonated hydroxylamide complex (which can
exist in the form of two valence tautomers as indicated in Figure 3).[15]
N2OR
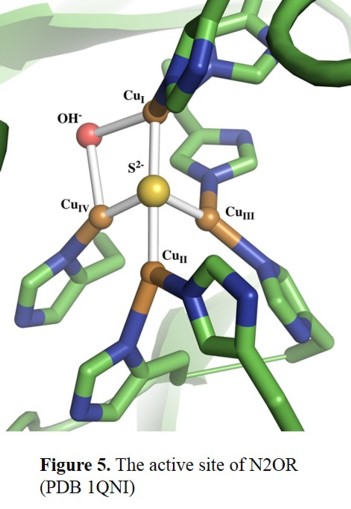
The crystal structure of N2O reductase (N2OR) has been determined for two different organisms. N2OR contains a unique copper
cluster, CuZ [16], which is the designated site of bonding and reduction of the N2O molecule. As shown in Figure 5,
the CuZ cluster consists of four copper centers, which are bridged by a sulfide ion. The cluster is connected to the protein
through seven His ligands that bind to the Cu centres. Additionally, the CuI-CuIV edge is bridged by a solvent-derived ligand (hydroxide?) in the P. nautica crystal structure,
and it has been proposed that this is the site of substrate binding.[17]
References:
D. J. Thomas, N. Lehnert
"The Biocoordination Chemistry of Nitric Oxide with Heme and Non-Heme Iron Centers";
in: 'Elsevier Reference Module in Chemistry, Molecular Sciences and Chemical Engineering';
Reedijk, J., Ed.,
Elsevier, 2017,
doi: 10.1016/B978-0-12-409547-2.11678-6
N. Lehnert, H. T. Dong, J. B. Harland, A. P. Hunt
"Reversing Nitrogen Fixation"
Nat. Chem. Rev. 2018, 2, 278-289
doi: 10.1038/s41570-018-0041-7
Literature:
[1] Ferguson, S. J. Curr. Opin. Chem. Biol. 1998, 2, 182-193.
[2] Richardson, D. J.; Watmough, N. J. Curr. Opin. Chem. Biol. 1999, 3, 207-219.
[3] Moura, I.; Moura, J. J. G. Curr. Opin. Chem. Biol. 2001, 5, 168-175.
[4] Hino, T.; Matsumoto, Y.; Nagano, S.; Sugimoto, H.; Fukumori, Y.; Murata, T.; Iwata, S.; Shiro, Y.
Science 2010, 330, 1666-1670.
[5] Zumft, W. J. Inorg. Biochem. 2005, 99, 194-215.
[6] Girsch, P.; de Vries, S. Biochim. Biophys. Acta 1997, 1318, 202-216.
[7] Praneeth, V. K. K.; Näther, C.; Peters, G.; Lehnert, N. Inorg. Chem. 2006, 45, 2795-2811.
[8] Berto, T. C.; Xu, N.; Lee, S. R.; McNeil, A. J.; Alp, E. E.; Zhao, J.; Richter-Addo, G. B.; Lehnert, N. Inorg. Chem. 2014, 53, 6398-6414.
[9] Blomberg, M. R. A. Biochemistry 2017, 56, 120-131.
[10] Speelman, A. L.; Zhang, B.; Silakov, A.; Skodje, K. M.; Alp, E. E.; Zhao, J.; Hu, M. Y.; Kim, E.; Krebs, C.; Lehnert, N. Inorg. Chem. 2016, 55, 5485-5501.
[11] Daiber, A.; Shoun, H.; Ullrich, V. J. Inorg. Biochem. 2005, 99, 185-193.
[12] Park, S.-Y.; Shimizu, H.; Adachi, S.-I.; Nakagawa, A.; Tanaka, I.; Nakahara, K.; Shoun, H.; Obayashi, E.; Nakamura, H.; Iizuka, T.; Shiro, Y.
Nature Struct. Biol. 1997, 4, 827-832.
[13] Shiro, Y.; Fujii, M.; Iizuka, T.; Adachi, S.-I.; Tsukamoto, K.; Nakahara, K.; Shoun, H.
J. Biol. Chem. 1995, 270, 1617-1623.
[14] Lehnert, N.; Praneeth, V. K. K.; Paulat, F. J. Comput. Chem. 2006, 27, 1338-1351.
[15] McQuarters, A. B.; Wirgau, N. E.; Lehnert, Curr. Op. Chem. Biol. 2014, 19, 82-89.
[16] Brown, K.; Tegoni, M.; Prudencio, M.; Pereira, A. S.; Besson, S.; Moura, J. J. G.; Moura, I.; Cambillau, C.
Nat. Struct. Biol. 2000, 7, 191-195.
[17] Chen, P.; Gorelsky, S. I.; Ghosh, S.; Solomon, E. I. Angew. Chem. Int. Ed. 2004, 43, 4132-4140.
|
