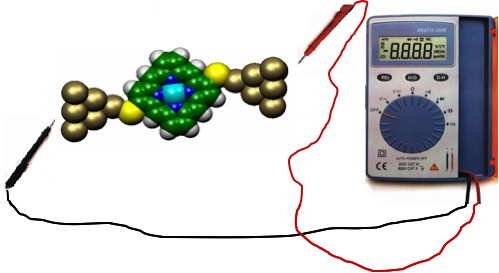
Aaron Christopher Tan , Janakiraman Balachandran , Seid Hossein Sadat , Vikram Gavini , Barry D. Dunietz , Sung-Yeon Jang , and Pramod Reddy
We present a combined experimental and computational study that probes the thermoelectric and electrical transport properties of molecular junctions. Experiments were performed on junctions created by trapping aromatic molecules between gold (Au) electrodes. The end groups (-SH,-NC) of the aromatic molecules were systematically varied to study the effect of contact coupling strength and contact chemistry. When the coupling of the molecule with one of the electrodes was reduced by switching the terminal chemistry from -SH to -H, the electrical conductance of molecular junctions decreased by an order of magnitude, whereas the thermopower varied by only a few percent. This observation, which provides information about the effect of contact coupling on the electronic structure of the junctions, has been predicted computationally in the past and is experimentally demonstrated for the first time. Further, our experiments and computational modeling indicate the prospect of tuning thermoelectric properties at the molecular scale. In particular, the thiol terminated aromatic molecular junctions revealed a positive thermopower that increased linearly with length. This positive thermopower is associated with charge transport primarily through the highest occupied molecular orbital (HOMO) as shown by our computational results. In contrast, a negative thermopower was observed for a corresponding molecular junction terminated by an isocyanide group due to charge transport primarily through the lowest unoccupied molecu- lar orbital (LUMO).
Nikolai Sergueev, Seungha Shin, Massoud Kaviany and Barry D. Dunietz,
The electron-phonon interaction is the dominant mechanism of inelastic scattering in molecular junctions. Here we report on its effect on the thermoelectric properties of single-molecule devices. Using density functional theory and the nonequilibrium Green's function formalism we calculate the thermoelectric figure of merit for a biphenyl-dithiol molecule between two Al electrodes under an applied gate voltage. We find that the effect of electron-phonon coupling on the thermoelectric characteristics strongly varies with the molecular geometry. Two molecular configurations characterized by the torsion angles between the two phenyl rings of 30deg and 90deg exhibit significantly different responses to the inelastic scattering. We also use molecular dynamics calculations to investigate the torsional stability of the biphenyl-dithiol molecule and the phonon thermal transport in the junction.
Heidi Philips, Alex Prociuk and Barry D. Dunietz,
The effect of bias and geometric
breaking on the
electronic spectrum of a model molecular system is studied.
Geometric symmetry breaking can either enhance the dissipative
effect of the bias, where spectral peaks are disabled, or
enable new excitations that are absent under zero bias
conditions. The analysis is performed on a simple model system by
solving for the electronic response to a pulse
perturbation in the dipole approximation. The dynamical response is extracted from the electronic
equations of motion as expressed by the Keldysh formalism. This
expression provides for the accurate treatment of an evolving
bulk-coupled system at the model Hamiltonian level.
Bei Ding, Victoria Washington and Barry D. Dunietz,
Transport properties of a Ni bis-&eta ² complex ligated by pairs of
bi-pyridazino derivatives are considered. This complex provides the
opportunity to avoid perpendicular alignment of the ligand $\pi$
planes. We study the effects of &pi bonding and of intramolecular
hydrogen bonding between the ligands as mediated by the metal center
on electron transport.
The complicated effect of the electronic structure
equilibration with the electrodes on the transport is discussed.
The analysis at the electronic structure level
provides guidelines to design a molecular bridge that is based on
metal complexation with effective electronic transport.
Alex Prociuk and Barry D. Dunietz,
The study of current induced by photoradiating a molecular-based device under bias is of fundamental importance to the improvement of photoconductors and photovoltaics. In this technology, electron pumps generate an uphill current that opposes a potential drop and thereby recharges a fuel cell. While the modeled molecular electron pump is completely symmetric, the sign of the photocurrent is solely determined by the existing bias and the nature of photoinduced electronic excitations. The photoradiation induces nonequilibrium population of the electrode-coupled system. The dependence of the photocurrent on electrode coupling, photoradiation field strength, and applied bias are studied at a basic model level.
Trilisa Perrine and Barry D. Dunietz,
The geometric aspects for the functionality of a molecular-based field
effect transistor (FET) are analyzed. A computational study is
performed on molecular models involving a well defined conjugation
plane coupled to gold-based electrodes through thiol
bonding. Transport gating of the FET is shown to depend on a
symmetry-breaking effect induced by the gating-field. This effect is
also related to the orientation of the field relative to the
gold-thiol bonds, the molecular conjugation plane, and the overall
symmetry of the device. It is found that the presence of a center of
inversion in the bulk-coupled molecular system results in the
cancellation of the transisting response. The presence of a plane of
symmetry which includes the transport vector, in a reduced symmetry
system, results in a gating response only to electric fields oriented
perpendicular to that mirror plane.
Alex Prociuk, Heidi Phillips and Barry D. Dunietz,
Non-equilibrium Green's function formalism (NEGF) by employing
time-dependent perturbation theory is used to solve the electronic
equations of motion of model systems under potential biasing
conditions. The time propagation is performed in the full frequency
domain of the two time variables representation. We analyze
transient aspects of the conductance under applied direct-current
and alternating current potential perturbation. The coherence
induced response dependence on different aspects of the applied
perturbation are resolved in time and analyzed using TD distribution
of the current operator.
Carlos R. Baiz, Sarah J. Ledford, Kevin J. Kubarych and Barry D. Dunietz
Conjugation effects on the thermodynamics of ground-state and lowest-singlet excited-state double hydrogen-atom transfer reactions in 7-azaindole and related models are studied with ab initio electronic structure methods. The results indicate that the extended conjugation of the system has a large effect on the relative energies required for hydrogen-atom transfer. The observed energy differences are mainly attributed to stabilization of the tautomer species by enhancing low-energy resonance structures and by allowing for efficient delocalization of excess charge in the reaction center.
Alex Prociuk and Barry D. Dunietz,
"Progress in Theoretical Chemistry and Physics" series book chapter in: Atomic and Molecular Systems, Dynamics, Spectroscopy,
Clusters, and Nanostructures, Springer publisher, 20 (2009) 265-277
The linear response of the electronic density of a molecular-based
junction under potential bias conditions to a probing polarizing
perturbation is calculated to model the electronic spectra. It is
shown that steady flux conditions lead to dramatic effects on the
electronic spectra of the confined system. The non-equilibrium
conditions enable electronic transitions that are otherwise forbidden.
The implemented methodology uses the Keldysh contour formalism to
express the electronic equations of motion. The related time
correlation Green Functions are then solved for in the full frequency
representation and at the linear response level.

Multiadsorption and Coadsorption of Hydrogen on Model Conjugated Systems
Miguel Wong, Benjamin Van-Kuiken, Corneliu Buda and Barry D Dunietz
J. Phys. Chem. C.,
113 (2009) 12571-12579.
Hydrogen interaction with conjugated aromatic molecular systems is
analyzed with high level ab initio electronic structure
methodology. The adsorption of hydrogen molecules on aromatic systems
ranging from a single ring to double ring systems including fused and
bridged molecules is investigated. We propose several
functionalization schemes of the conjugated system to increase
hydrogen intake. These functionalizations consist of using an
asymmetrical conjugation skeleton as inherently present for the
azulene molecule and the introduction of B�$(B!]�(BN heteroatom pairs as
doping sites in the conjugation ring. We have also tested the
possibility of involving more than a single interaction site within
the same molecule by using a molecule involving several rings as the
triptycene and several derived functionalized molecules. These
computational studies provide insight on the interaction of hydrogen
with conjugated molecular species. This insight can be utilized for
designing materials such as metal organic frameworks or other porous
organic polymers with enhanced uptake properties.
Enhanced Conductance via Induced pi-Stacking Interactions in Cobalt(II) Terpyridine Bridged Complexes
Trilisa M. Perrine, Timothy Berto and Barry D. Dunietz
J. Phys. Chem. b.,
112 (2008) 16070-16075.
Computational model systems are used to explore improving the
transmission through a molecular device based on bridged cobalt(II)
complexes. The bridging ligands and the organic conjugated molecular
ligands are altered to improve the current flow through both an
enhanced �$(B&P�(B-stacking interaction as well as involving the metal ions
directly in the conduction pathway. With terpyridine as the organic
ligand, both acetate and NH2�$(B!]�(B produce conductive devices, while a
terpyridine complex bridged by Cl�$(B!]�(B is not conductive. The addition of
a fused ring on either end of the conjugated molecule has a complex
effect which is sensitive to the bridged ligand and the particular
geometry of the complex.
Time-dependent current through electronic channel models using a mixed time-frequency
solution of the equations of motion
Alexander Prociuk and Barry D. Dunietz
Phys. Rev. B,
78
(2008) 165112.
A non-equilibrium Green's-Function (NEGF) model based on time
dependent perturbation theory is developed to propagate electronic
structure and molecular conductance of extended
electrode-molecule-electrode nanostructures. In this model, we use
the two time variable nature of the Kadanoff-Baym equations of motion
to formulate a mixed time-frequency representation for the electronic
density expressed by the appropriate GF (G<). This allows for the
dynamical treatment of open systems. Furthermore, highly informative
time dependent Wigner distributions are used to shed light on the
features of dynamical observables, such as electron current.
Calculations, performed on model systems, resolve the dynamic current
into direct and alternating components. The direct current is due to
electronic open channels near the Fermi level and the alternating
response is due to interference fringes from a superposition of
extended states. We analyze the transient conductance with respect to
the fundamental system's parameters, the effect of bound states and
conductance driven by laser induced coherence affected by detuning due
to an applied DC bias. The amplitude of the alternating transient
current can be adjusted by reshaping the bias pulse or by controlling
the electronic coupling terms. Bound states may yield a persisting
oscillating response depending on their relative electronic densities.
In the analysis we utilize the calculated highly informative
time-dependent current distributions.
Ab initio study of charge transport of hydrogen functionalized palladium wires
Zhen Zhao, and Barry D. Dunietz, J. Chem. Phys, 129 (2008) 024702.
We present ab initio calculations of transport properties of palladium
wires in the presence of hydrogen. Detailed investigations have been
1conducted with a pure palladium wire and with opening a gap inside the
wire in which the transition between point contact regime and
tunneling regime occurs. The effect of the presence of hydrogen in the
gap is studied for different ranges of the gap size. The hydrogen
mediated transport in the contact and tunneling regimes of the gap are
analyzed and compared. It is predicted that only in large enough
distances the hydrogen presence increases the conductance. The effect
of additional hydrogen molecules on the gap is also studied.
Synthetic, mechanistic, and computational investigations of nitrile-alkyne cross-metathesis
Andrea M. Geyer, Eric S. Wiedner, J. Brannon Gary, Robyn L. Gdula, Nicola C. Kuhlmann, Marc J. A. Johnson, Barry D. Dunietz, Jeff W. Kampf
J. Amer. Chem. Soc. , 130 (2008) 8994-8999.
The terminal nitride complexes NW(OC(CF3)2Me)3(DME) (1-DME), [Li(DME)2][NW(OC(CF3)2Me)4] (2), and [NW(OCMe2CF3)3]3 (3) were prepared in good yield by salt elimination from [NWCl3]4. X-ray structures revealed that 1-DME and 2 are monomeric in the solid state. All three complexes catalyze the cross-metathesis of 3-hexyne with assorted nitriles to form propionitrile and the corresponding alkyne. Propylidyne and substituted benzylidyne complexes RCW(OC(CF3)2Me)3 were isolated in good yield upon reaction of 1-DME with 3-hexyne or 1-aryl-1-butyne. The corresponding reactions failed for 3. Instead, EtCW(OC(CF3)Me2)3 (6) was prepared via the reaction of W2(OC(CF3)Me2)6 with 3-hexyne at 95 �N0C. Benzylidyne complexes of the form ArCW(OC(CF3)Me2)3 (Ar = aryl) then were prepared by treatment of 6 with the appropriate symmetrical alkyne ArCCAr. Three coupled cycles for the interconversion of 1-DME with the corresponding propylidyne and benzylidyne complexes via [2 + 2] cycloaddition�$(B!]�(Bcycloreversion were examined for reversibility. Stoichiometric reactions revealed that both nitrile-alkyne cross-metathesis (NACM) cycles as well as the alkyne cross-metathesis (ACM) cycle operated reversibly in this system. With catalyst 3, depending on the aryl group used, at least one step in one of the NACM cycles was irreversible. In general, catalyst 1-DME afforded more rapid reaction than did 3 under comparable conditions. However, 3 displayed a slightly improved tolerance of polar functional groups than did 1-DME. For both 1-DME and 3, ACM is more rapid than NACM under typical conditions. Alkyne polymerization (AP) is a competing reaction with both 1-DME and 3. It can be suppressed but not entirely eliminated via manipulation of the catalyst concentration. As AP selectively removes 3-hexyne from the system, tandem NACM-ACM-AP can be used to prepare symmetrically substituted alkynes with good selectivity, including an arylene-ethynylene macrocycle. Alternatively, unsymmetrical alkynes of the form EtCCR (R variable) can be prepared with good selectivity via the reaction of RCN with excess 3-hexyne under conditions that suppress AP. DFT calculations support a [2 + 2] cycloaddition�$(B!]�(Bcycloreversion mechanism analogous to that of alkyne metathesis. The barrier to azametalacyclobutadiene ring formation/breakup is greater than that for the corresponding metalacyclobutadiene. Two distinct high-energy azametalacyclobutadiene intermediates were found. These adopted a distorted square pyramidal geometry with significant bond localization.
Accessing Metal-Carbide Chemistry. A Computational Analysis of Thermodynamic Considerations
J. Brannon Gary, Corneliu Buda, Marc J. A. Johnson, and Barry D. Dunietz, Organometallics, 27 (2008) 814.
The electronic structures of terminal metal carbide complexes are
calculated using DFT. This study outlines the factors that give rise
to stable carbide complexes, which can be used to help in the
synthesis of new carbide complexes and to tune their stability as
desired. The calculations reveal the presence of a strong Ru&equiv C triple
(&sigma + 2&pi) bond. The C atom is nearly unhybridized, such that the
C-component of the Ru-C \sigma-bond has 90% 2p character. This leaves a
very stable carbon-based lone pair that is almost entirely 2s in
character, which accounts for the lack of Lewis base character
exhibited experimentally. Calculations predict a Ru-C bond
dissociation energy of 147.4 kcal mol^{-1} in a typical Ru carbide
complex. This large bond strength is not unique to the RuC bond, as
revealed by an extension of the study to identify schemes by which to
chemically tune the metal-carbide bond strength. Methods examined to
achieve this tuning include changing the identity of the central metal
and altering the metal ligation scheme. In general, 16-electron
square-pyramidal M(C)L_4 complexes and 12- or 16-electron tetrahedral
M(C)L_3 complexes of the 4d elements can possess comparably strong
metal-carbide bonds. The calculations also show that the carbide
moiety exerts a very strong trans influence, which explains several
experimental observations. We conclude that the dearth of terminal
carbide complexes is not due to any inherent weakness of M&equiv C
bonds. Many more terminal carbide complexes can be expected in the
future as new routes to their formation are found.
Gating of single molecule transistors: Combining field-effect and chemical control
Trilisa M. Perrine, Ron G. Smith, Christopher Marsh, and Barry D. Dunietz, Journal of Chemical Physics, 128 (2008) 154706.
Previously we have demonstrated that several structural features
are crucial for the functionality of molecular field effect transistors.
The effect of additional
structural aspects of molecular wires is explored.
These include the type of, the thiol binding location on, and the chemical substitutions of a conjugated
system. Pentacene, porphyrin and the Tour-Reed devices are utilized as model
systems.
The thiol binding location is shown to have a varried effect on the transmission
of a system depending on the molecular orbitals involved.
Substitution by electron withdrawing and donating groups is illustrated
to have a substantial effect on the transmission of single molecule devices.
The substituation effect is either a simple energy shifting effect
or a more complicated
resonance effect, and can be used to effectively tune the electronic behavior of a single molecule field effect transistor.
Conductance of a cobalt(II) terpyridine complex based molecular transistor: A computational analysis
Trilisa M. Perrine and Barry D. Dunietz, Journal of Physical Chemistry A: Lester Special Issue, 112 (2008), 2043-2048.
A recent experiment, in which a molecular transistor based on the coordination chemistry of cobalt(II) and organic self-assembly-monolayers is formed by means of self-aligned lithography, is analyzed with a computational approach. The calculations reveal that a complex involving two cobalt(II) ions bridged by acetate ions can effectively span the nano-gap. This bridged complex is shown to be both more flexible and more conductive than the alternative structure involving a
single cobalt(II) ion. The single cobalt(II) ion complex is the more stable structure in a non-confined environment (i.e. in solution), but is found to be less effective at connecting the leads of the fabricated gap and is less
likely to result in a conductive device.
Carbonyl mediated conductance through metal bound peptides; a computational study
Trilisa M. Perrine and Barry D. Dunietz, Nanotechnology, 18, (2007), 424003.
Large increases in the conductance of peptides, upon binding to metal ions, have been recently reported experimentally. The mechanism of the conductance switching is examined computationally. It is suggested that oxidation of the metal ion occurs after binding peptide. This is caused by the bias potential placed across the metal-peptide complex. A combination of configurational changes, metal ion involvment and interactions between carbonyl group oxygen atoms and the gold leads are all shown to be necessary for the large improvement in the conductance seen experimentally. Differences in the molecular orbitals of the nickel and copper complexes are noted and serve to explain the variation of the conductance improvement upon binding to either a nickel or copper ion.
Theoretical Studies of Conjugation Effects on Excited State Intramolecular Hydrogen-atom
Transfer Reactions in Model Systems
Carlos Baiz and Barry D. Dunietz, Journal of Physical Chemistry A, 111, (2007), 10139-10143.
Intramolecular Hydrogen-atom transfer dependence on electronic
conjugation of curcumin and related molecular models in the ground
state and 1&pi &pi* excited state are computationally studied at
first-principles electronic structure level. The larger, more
conjugated, systems exhibit a lower reaction barrier in the ground
state but a higher barrier in the excited state. This is associated
with a smaller increase in the conjugation upon excitation in the
larger systems. Our studies provide a detailed description and
analysis of these energy trends as well as an insight into the
physical nature of the intramolecular hydrogen-atom transfer
reactions.
Fragmentation pathways and mechanisms of aromatic compounds in atmospheric pressure studied by GC-DMS and DMS-MS
Shai Kendler, Gordon R. Lambertus, Barry D. Dunietz, Stephen L. Coy, Erkinjon G. Nazarov, Raanan A. Miller, and Richard D. Sacks, Int. J. of Mass Spec. , 263, (2007), 137-147.
Differential mobility spectrometry (DMS) is a highly sensitive sensing technology capable of selecting and detecting ions based on the difference between ion mobility at high and low electric field. The combination of a micro-fabricated DMS with gas chromatography (GC) has allowed extensive investigation of the ion chemistry and collisionally induced dissociation (CID) of diaryl molecules on a millisecond timescale at temperatures up to 130 degrees C. DMS-pre-filtered time-of-flight mass spectrometry (DMS-MS) has been used to verify the chemical composition of the ion species resolved by GC-DMS. This work focuses on the fragmentation of diaryl compounds, including diphenyl methane (DPM) and bibenzy] (BB), using information from the DMS and DMS-MS spectra of a series of aromatic compounds. Density functional theory calculations have been used to investigate the geometry and the energy along the reaction coordinate for the loss of benzene from DPM-H+ and BB-H+ for comparison with GC-DMS and DMS-MS experimental results and with previously reported chemical ionization MS. DPM-H+ is observed to undergo field-induced fragmentation in the DMS to produce C7H7+(Bz(+)) and unobserved neutral benzene with a low energy barrier. In contrast, BB-H+ fragments to C8H9+ and benzene with a higher energy barrier. Calculated barriers and experimental results are in qualitative agreement. Depletion of the ionized fragments in favor of ion-neutral clusters was also observed at higher concentrations. It is suggested that CID in DMS can further enhance DMS analytical performance.
Single-molecule field-effect transistors: A computational study of the effects of contact geometry and gating-field orientation on conductance-switching properties
Trilisa M. Perrine and Barry D. Dunietz, Phys. Rev. B, 75, (2007), 195319.
The relation of geometric features to the effect of gating electric fields on the conductance through conjugated systems is investigated by electronic transmission calculations employing Green's function based modeling. Switching is only induced if the field is applied in an orientation which results in energy shifting of the molecular orbitals. This is found to depend on the orientation of the field with respect to the plane defined by the molecular conjugation. The switching can be quenched by structural rearrangement of the chemical bonds to the bulk, where the relative position of the electrodes is modified.
Electron Transport through Heterogeneous Intermolecular Tunnel Junctions
Das, M. and Dunietz, B. D., J. Phys. Chem. C., 111, (2007), 1535--1540.
Quantum charge transport through intermolecular tunnel junctions is studied. Intermolecular tunnel junctions can be defined by the end groups of pairs of self-assembled monolayers of functionalized conjugated alkenes on gold surfaces. Conductivity dependence on the tunnel distance has been compared for various junctions. It is found that for junctions dominated by attractive interactions, for example, systems involving hydrogen bonding, conductivity exhibits less dependence on the tunneling distance than with junctions dominated by dispersive interactions. Junctions with stronger distance dependence conductivty are desired for applications related to chemical sensors. Our study provides insight for designing an efficient chemical sensor that is based on heterogeneous tunnel junction, which may involve conductivity through a combination of the attractive hydrogen-bonding channel and repulsive dispersive interactions.
Benchmarking the performance of density functional theory based Green's function formalism utilizing different self-energy models in calculating electronic transmission through molecular systems
Prociuk, A. and Dunietz, B.D., J. Chem. Phys., 125, (2006), 204717.
Electronic transmission through a metal-molecule-metal system is calculated by employing a Green's function formalism in the scattering based scheme. Self-energy models representing the bulk and the potential bias are used to describe electron transport through the molecular system. Different self-energies can be defined by varying the partition between device and bulk regions of the metal-molecule-metal model system. In addition, the self-energies are calculated with different representations of the bulk through its Green's function. In this work, the dependence of the calculated transmission on varying the self-energy subspaces is benchmarked. The calculated transmission is monitored with respect to the different choices defining the self-energy model. In this report, we focus on one-dimensional model systems with electronic structures calculated at the density functional level of theory.
Spin-dependent electronic transport through a porphyrin ring ligating an Fe(II) atom: An ab initio study
Chen, Y. and Prociuk, A. and Perrine, T. and Dunietz, B.D., Phys. Rev. B, 74, (2006), 245320.
Conductance calculations employing density functional theory methodology and Landauer formalism predict that a ligated iron atom can be used as a switching device. The iron atom is ligated in our models by a porphyrin molecule. The iron-porphyrin molecular device is shown to lose more than 66% of its conductance by shifting from the low spin coupling state to excited spin states. Further reduction is also correlated with a mechanical distortion of the porphyrin plane. Both the distortions and spin transitions are fast processes that can be invoked by manipulating the iron's ligation scheme through the axial ligands.
Hydrogen Physisorption on the Organic Linker in Metal Organic Frameworks: Ab Initio Computational Study
Buda, C. and Dunietz, B.D., J. Phys. Chem. B., 110, (2006), 10479--10484.
Research for materials offering efficient hydrogen storage and transport has recently received increased attention. Metal organic frameworks (MOFs) provide one promising group of materials where several recent advances were reported in this direction. In this computational study ab initio methods are employed to study the physisorption of hydrogen on conjugated systems. These systems are used as models for the organic linker within MOFs. Here, we focus on the adsorption sites related to the organic linker with special attention to the edge site, which was only recently reported to exist as the weakest adsorbing site in MOFs. We also investigate chemically modified models of the organic connector that result in enforcing this adsorption site. This may be crucial for improving the uptake properties of these materials to the goal defined by DOE for efficient hydrogen transport materials.
Metathesis-Enabled Formation of a Terminal Ruthenium Carbide Complex: A Computational Study
Buda, C., Caskey, S.R., Johnson, M.J.A. and Dunietz, B.D., Org. Metal., 25, (2006), 4756-4762.
The energy profile of rare Ru carbide formation starting from an acetoxycarbene complex is studied using DFT methods. Three distinctive reaction pathways that differ in their initiation step are investigated. Two of the proposed reaction mechanisms have relatively similar activation barriers. Therefore, additional calculations have been performed using large size ligands (PCy3), matching exactly the actual experimental system. In addition, the corresponding kinetic isotope effect has been evaluated and compared to the experimental measured value.
Additional Manuscripts and Papers:
Advances in methods and algorithms in a modern quantum chemistry program package.
Shao, Yihan, Molnar, Laszlo Fusti, Jung, Yousung, Kussmann, Jorg, Ochsenfeld, Christian, Brown, Shawn T., Gilbert, Andrew T.B., Slipchenko, Lyudmila V., Levchenko, Sergey V., O'Neill, Darragh P., Jr, Robert A. DiStasio, Lochan, Rohini C., Wang, Tao, Beran, Gregory J.O., Besley, Nicholas A., Herbert, John M., Lin, Ching Yeh, Voorhis, Troy Van, Chien, Siu Hung, Sodt, Alex, Steele, Ryan P., Rassolov, Vitaly A., Maslen, Paul E., Korambath, Prakashan P., Adamson, Ross D., Austin, Brian, Baker, Jon, Byrd, Edward F. C., Dachsel, Holger, Doerksen, Robert J., Dreuw, Andreas, Dunietz, Barry D., Dutoi, Anthony D., Furlani, Thomas R., Gwaltney, Steven R., Heyden, Andreas, Hirata, So, Hsu, Chao-Ping, Kedziora, Gary, Khalliulin, Rustam Z., Klunzinger, Phil, Lee, Aaron M., Lee, Michael S., Liang, WanZhen, Lotan, Itay, Nair, Nikhil, Peters, Baron, Proynov, Emil I., Pieniazek, Piotr A., Rhee, Young Min, Ritchie, Jim, Rosta, Edina, Sherrill, C. David, Simmonett, Andrew C., Subotnik, Joseph E., III, H. Lee Woodcock, Zhang, Weimin, Bell, Alexis T. and Chakraborty, Arup K., Phys. Chem. Chem. Phys., 8, (2006), 3172-3191.
Articles Before 2005:
Ugalde, J. M., Dunietz, B., Dreuw, A., Head-Gordon, M. and Boyd, R. J. `The spin
dependence of spatial size of Fe(II) and of the structure of Fe(II)-porphyrins.' J. Phys. Chem. A., 108, (2004), 4653-4657.
Dunietz, B. D., Markovic, N., Ross, P. H. and Head-Gordon, M. `Initiation of Electrooxidation of CO on Pt based electrodes at full coverage conditions simulated by ab-initio electronic structure calculations.' J. Phys. Chem. B., 108, (2004), 9888.
Saravanan*, C., Dunietz*, B. D., Markovic, N., Somorjai, G. and Head-Gordon,
M. Ross, P. H. `Electro-oxidation of CO on Pt electrodes simulated by electronic structure calculations.' J.Electroanal.Chem., J. Weaver Special memorial issue (v554), (2003), 459.
Dunietz, B. D. and Head-Gordon, M. `Manifestations of symmetry breaking in selfconsistent eld electronic structure calculations.' J. Phys. Chem. A., 107, (2003), 9160.
Head-Gordon, M., Van Voorhis, T., Beran, G. J. O. and Dunietz, B. D. `Local correlation models.' Computational Science - ICCS 2003, Pt IV , 2660, (2003), 96-102.
Dunietz, B. D., Dreuw, A. and Head-Gordon, M. `Initial steps of the photodissociation of the CO ligated heme group.' J. Phys. Chem. B., 107, (2003), 5623-5629.
Dunietz, B. D., van Voorhis, T. and Head-Gordon, M. `Geometric direct minimization of Hartree Fock calculations involving open shell wavefunctions with spin restricted orbitals.' J. Theo. and Comp. Chem., 1, (2002), 255-261.
Dreuw, A., Dunietz, B. D. and Head-Gordon, M. `Characterization of the relevant
excited states in the photodissociation of the CO-ligated Hemoglobin and Myoglobin.'
J. Am. Chem. Soc., 124, (2002), 12070-12071.
Dunietz, B. D. and Friesner, R. A. `Application and development of multicongurational localized perturbation theory.' J. Chem. Phys., 115, (2001), 11052.
Friesner, R. A. and Dunietz, B. D. `Large-scale ab-initio quantum chemical calculations on biological systems.' Accounts Chem Res, 34, (2001), 351-358.
Gherman, B. F., Dunietz, B. D., Whittington, D. A., Lippard, S. J. and Friesner, R. A. `Activation of the C-H bond of methane by intermediate Q of methane monooxygenase:
A theoretical study.' J. Am. Chem. Soc., 123, (2001), 3836.
Dunietz, B. D., Beachy, M. D., Cao, Y. X., Whittington, D. A., Lippard, S. J. and
Friesner, R. A. `Large scale ab-initio quantum chemical calculation of the intermediate
in the soluble methane monooxygenase catalytic cycle.' J. Am. Chem. Soc., 122, (2000), 2828.
Friesner, R. A., Murphy, R. B., Beachy, M. D., N., Ringnalda M., Pollard, W. T.,
Dunietz, B. D. and Cao, Y. X. `Correlated ab-initio electronic structure calculations
for large molecules.' J. Phys. Chem. A., 103, (1999), 1913.
Dunietz, B. D., Murphy, R. B. and Friesner, R. A. `Calculation of atomization energies by a multicon gurational localized perturbation theory - Application for closed shell cases.' J. Chem. Phys., 110, (1999), 1921.